Introduction
Whereas the holy grail of Artificial Intelligence (AI) research is to create General AI (GAI), the ultimate aspirational goal of human intelligence research is to increase general intelligence (g) in individuals (Haier, 2023). To accomplish this, collaborative efforts can build on 120 years of well-replicated psychometric research and recent discoveries in genetics, neuroimaging, and cognitive neuroscience. We review key findings from these fields that may stimulate excitement for molecular biology studies of intelligence similar to those that focus on learning and memory, both important cognitive components of the g-factor. Often molecular research on them is funded for their potential for cognitive enhancement in patients with dementia or other brain damage.
This paper is organized to review recent findings relevant to causal questions about the molecular brain and differences in g among individuals. The statistical existence of the g-factor itself, and its correlations to a wide variety of socially important outcomes and brain measures, are possibly the most replicated findings in all of psychology research (Haier et al., 2024; Warne, 2020), including cross-cultural studies (Warne & Burningham, 2019).
Neuroimaging and cognitive neuroscience have identified specific structurally-implicated functional neural networks related to psychometric intelligence, especially the Parietal-Frontal Network (Basten et al., 2015; Hilger et al., 2017; Jung & Haier, 2007; Thiele et al., 2022) and the Default Mode Network (DeSerisy et al., 2021; Song et al., 2009; Yadav & Purushotham, 2025). One brain area of particular interest in these networks is the precuneus in the parietal lobe. The precuneus shows both the greatest expansion relative to non-human primates and the highest resting metabolic demands of any cortical region—suggesting that its growth was a high-cost, high-benefit adaptation supporting introspective planning, and integrative cognition, all heavily loaded on general intelligence (g), and associated with the sophisticated capacity to use tools and manufacture complex objects (Bruner, 2023; Bruner et al., 2017).
Genetic discoveries are also beginning to suggest causal mechanisms for individual differences in g (J. Lee & Morris, 2025). For example, polygenic risk scores (PRS) generated by genome-wide association studies (GWAS) indicate that many genes have tiny additive influences, with very few major-effect loci. These findings, generally well-replicated, provide hints about causality that have led to speculations about brain efficiency, metabolism, mitochondria, and many other brain processes and mechanisms (Deary et al., 2022; Geary, 2018; Haier, 2023).
The rest of this paper provides background for psychometricians and neuroimaging researchers who want to seek collaborations with molecular researchers. Looking across these findings, we end by suggesting collaborations to address questions about specific gene expressions, neuronal functions, brain pathways, and cognitive performance related to intelligence (Moodie et al., 2023).
Psychometric and neuroimaging background
Readers of Intelligence and Cognitive Abilities (ICA) are familiar with well-replicated research that defines the g-factor as the main empirical component of intelligence (Coyle, 2021; A. R. Jensen, 1998) across cultures (Warne & Burningham, 2019). There are different psychometric and cognitive theories about what the g-factor reflects (Haier et al., 2024) and why it correlates to a wide variety of socially important variables. Since the early neuroimaging studies of intelligence (Haier et al., 1988; Jung & Haier, 2007), these theories now include putative brain mechanisms based on lesion mapping (A. K. Barbey et al., 2012, 2013) and sophisticated connectome studies (A. Barbey, 2018, 2021; A. Barbey et al., 2021; Fraenz et al., 2021; Hilger & Sporns, 2021). Of particular interest, for example, the PFIT network and the Default Mode Network have been related to individual differences in psychometric measures of intelligence (Basten et al., 2015; DeSerisy et al., 2021; Hilger et al., 2017; Jung & Haier, 2007; Song et al., 2009; Thiele et al., 2022; Yadav & Purushotham, 2025); (DeSerisy et al., 2021; Song et al., 2009; Yadav & Purushotham, 2025). Both networks feature association areas of the cortex and the precuneus. There is an emerging area of research focused on cognitive network neuroscience (A. Barbey, 2018). These brain network findings have been extended to studies of neuronal and dendritic levels (Genc et al., 2018; Goriounova et al., 2018; Goriounova & Mansvelder, 2019; Thiele et al., 2023). However, as is typical, there are inconsistent findings and no compelling weight-of-evidence indicates strong causal relationships between brain assessments and individual differences in psychometric intelligence measures although there are some promising studies (J. J. Lee et al., 2019; Schubert et al., 2018). Experiments that aim to manipulate brain network function using Transcranial Magnetic Stimulation (TCMS), drugs, and other procedures are in early stages but they may advance understanding causal mechanisms (Neubauer et al., 2017; Schubert et al., 2018). Such studies may become the next phase of intelligence research collaborations among psychometricians, cognitive neuroscientists, and neuroimaging researchers. We believe that developments in genetic research can provide even more opportunities for collaborations that may identify causal molecular influences on intelligence.
Molecular genetic research and intelligence
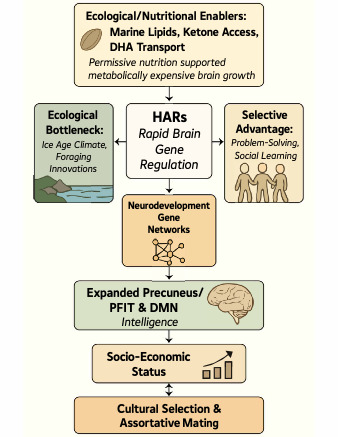
The extent to which genes influence individual difference in intelligence, especially the g-factor, is not yet conclusive since strong heritability estimates from decades of behavioral genetic studies have not yet been found in GWAS studies (Oxley et al., 2024; Plomin & von Stumm, 2018). In our view, it is likely that these inconsistencies will be sorted out with additional research (despite some gloomy doubts) but evolution and brain development are key to understanding intelligence on a molecular level. In this paper we present an overview on why we think this is so based on specific molecular genetic findings (see Figure 1 about here).
__70k_years_ago.png)
Human Accelerated Regions (HARs): The chimpanzee, our closest extant relative, diverged about 6-7 million years (Myr) ago and shares about 96% overall genetic identity, and a remarkable 98.8% identity in coding exons (the sections of a gene that contain instructions for building proteins). Gorillas diverged just prior to the chimps, about 8-10 Myr ago and share about 93% identity while “Lucy” (Australopithecus afarensis) who diverged about 3-4 Myr ago is estimated to have shared about 97% identity; and the Neanderthals, diverged only about 700 thousand years ago sharing 99.88% (Consortium, 2005; Green et al., 2010; Patterson et al., 2006; Scally et al., 2012).
Divergence percentages typically reflect neutral mutations that likely underrepresent evolution-selected changes, which in regulatory regions might drive development of new human-specific traits. Such selected regions are Human Accelerated Regions (HARs). They are the tiny segment of the genome, about 0.03%, that distinguishes modern man from our relatives (Pollard et al., 2006; Prabhakar et al., 2006; Whalen & Pollard, 2022). This approximately 1.5Mb is over 95% non-coding DNA and it is distributed as sequences of 100-300bps found adjacent to, apparently regulating, genes crucial for neurodevelopment, including those associated with autism spectrum disorder (ASD), intellectual disability (ID), and schizophrenia (Doan et al., 2016; Won et al., 2019). They appear to be enhancers that have undergone accelerated substitution rates in humans, suggesting positive selection and they are expressed early during brain development with some having been shown to explicitly lead to increased brain size by accelerating neural progenitor proliferation during cortical development. Brain expansion has largely been in networks subserving higher-order functions, increasing cognitive properties that are highly heritable and relate to genetic effects associated with neuron growth and metabolism (Capra et al., 2013; Uebbing et al., 2021; Zhao et al., 2021). Uncovering to what extent cognitive functional networks have developed in recent human evolution, is crucial for our understanding the high cognitive complexity of human brain function, an important locus of genetic changes in human brain evolution.
HAR activity is particularly pronounced in the association cortex and the precuneus, areas that are amongst the brain structures displaying the highest resting metabolic rates (Cavanna & Trimble, 2006) and areas involved in the PFIT and Default Mode networks. Notably, several HARs remain polymorphic in modern populations, confirming their recent and ongoing evolution, suggesting they are not yet fixed alleles and may still be under selective pressure (Uebbing et al., 2021). GWAS of socioeconomic status (SES) and educational attainment (EA) indeed highlight loci and genes that overlap significantly with HARs (Driessens et al., 2023; J. J. Lee et al., 2018).
Moreover, genes that correlate in their expression levels with dendritic length and action potential kinetics—traits linked to intelligence—are enriched within the intersection of HAR, IQ, and EA gene sets, and are involved in synaptic functions (Driessens et al., 2023; Goriounova et al., 2018). This points to a functional overlap of both SES/EA-associated GWAS genes and HAR-linked genes converging on neuron growth, connectivity, and function in brain areas tied to cognition. Moreover, many of these loci map to genes involved in neural development, synaptic function, and brain structure —processes also under HAR regulation (Driessens et al., 2023). Educational attainment (EA) GWAS (Davies et al., 2015; Doan et al., 2016; J. J. Lee et al., 2018; Won et al., 2019) have identified autosomal loci associated with years of schooling (Okbay et al., 2016) and an Income GWAS (N ≈ 668,000) found 162 loci tied to a general SES factor, showing very high genetic correlation with EA (r ≈ 0.92) (Kweon et al., 2025).
While direct SNP-by-SNP overlaps between these EA/SES/IQ GWAS and HAR GWAS aren’t often reported, gene-set enrichment studies show EA/IQ-associated genes frequently fall within HAR topological domains/enhancer-interacting regions and shared pathways include neurogenesis, synaptogenesis, neuronal projection development, and energy metabolism—all enriched among HAR-proximal genes (Kweon et al., 2025; Okbay et al., 2016, 2022). Therefore, GWAS of SES and EA identify hundreds of loci and genes tied to cognitive function and many of these genes overlap with HAR-associated genes, converging at the level of neuronal structure (dendritic complexity, connectivity), electrical function (action potential speed), synapse biology (plasticity, neurotransmission) and cortical association regions (Driessens et al., 2023). This overlap is statistically robust at the gene-set and pathway level, implying HAR-regulated elements, those genomic elements that most specifically define modern man, contribute to the genetic architecture of SES/EA and cognitive ability. Given the astronomical number of neurons (about 100 billion) and neuronal connections (about 100 trillion) in the human brain (Lent et al., 2012) even the slightest changes in the efficiency of single neuron function can translate into large differences in the potential for signal/information processing (i.e. cognition).
Cognition and Molecular Genetics
The disproportionate expansion of association cortices, particularly the precuneus, marks a hallmark of recent hominin brain evolution. This region, central to the PFIT and the DMN, is involved in social cognition, memory and self-aware agency, and interpreting visuo-spatial imagery from the first-person perspective (Kjaer et al., 2002; Schroeter et al., 2023). It is characterized by transient decreases in tonic activity during engagement in non-self-referential goal-directed actions, all uniquely human cognitive capacities (Cavanna & Trimble, 2006). Damage to the precuneus in patients leads to impairments in working memory, visuospatial reasoning, episodic memory—cognitive domains which heavily load onto g.
The precuneus shows both the greatest expansion relative to non-human primates and the highest resting metabolic demands of any cortical region—suggesting that its growth was a high-cost, high-benefit adaptation supporting introspective planning, and integrative cognition. Recent studies link this anatomical expansion of the brain to specific rapid genomic changes: HARs (Doan et al., 2016; Pollard et al., 2006; Won et al., 2019). These rapidly evolved noncoding sequences that function as enhancers during fetal brain development, preferentially regulate genes expressed in the DMN, particularly the precuneus. Bruner and colleagues (Bruner & Iriki, 2016) directly compared midsagittal MRI templates of humans and chimpanzees and found that the most pronounced inter-species shape difference is a “longitudinal spatial dilation of the upper part of the precuneus” in humans, with no overlap in morphological range between the two species.
This work establishes the precuneus as the single cortical region with the greatest phylogenetic expansion. PET/fMRI studies of the Default Mode Network (DMN) routinely show that among all cortical areas, the posterior precuneus/posterior cingulate cortex exhibits the largest baseline glucose-metabolic response at rest. In particular, (Gusnard et al., 2001; Utevsky et al., 2014) demonstrated that the precuneus has the highest metabolic response during rest of any DMN node. This finding has been replicated across modalities and cohorts, marking the precuneus’ ionic and synaptic baseline as the most energetically costly to maintain. HARs are strongly over-represented in DNase-hypersensitive sites and histone-marked enhancers active prenatally, with the greatest enrichment in fetal brain compared to other tissues (Won et al., 2019). This confirms that many HARs function as developmental enhancers shaping early cortical neurogenesis. Comparative transcriptomics show that genes linked to HARs are most highly expressed in higher-order cognitive networks, with the Default Mode Network (DMN) exhibiting the strongest HAR-gene signature, peaking in regions like the precuneus (Wei et al., 2019). It would be of interest if neuroimaging research seeded parts of the precuneus for connectivity studies that might identify brain networks more strongly correlated with intelligence than the PFIT or the DMN.
Functional assays show that many HARs drive gene expression in radial glia and excitatory neurons of cortical layer II/III—precisely where recent IQ-linked proteomic signatures (Cui et al., 2025; Zhang et al., 2025) and neuronal morpho-electric signatures (Goriounova et al., 2018) are most prominent. This convergence is deepened by transcriptomic and proteomic data: Zhang et al. identified 44 proteins whose levels in dorsolateral prefrontal cortex (dPFC) were associated with IQ. Many of these are cis-regulated by loci also implicated in GWAS of educational attainment (EA). The most significant proteins, such as those involved in synaptic function, dendritic growth, and mitochondrial activity, mirror the biological processes governed by HAR-regulated networks.
Taken together, these findings—from enhancer assays in neuronal progenitor cells (NPCs) and neurons, in vivo HAR knock-ins, to single-cell layer-specific expression maps and IQ-linked proteomics—converge on a coherent picture: HARs drove the evolution of regulatory programs in radial glia and layer II/III excitatory neurons, the very cells where IQ-associated proteins are most abundantly expressed. This compelling multi-modal evidence is internally consistent across genomic, epigenomic, transcriptomic, and proteomic levels. Crucially, these genes linked to HARs, IQ, and EA are not randomly distributed, but are highly co-expressed in human L2/L3 cortical pyramidal neurons—cells that form the substrate for the long-range associative connectivity in the precuneus and other DMN hubs (Driessens et al., 2023; Krienen et al., 2020). These neurons also exhibit unique properties such as long dendrites and faster action potentials; traits linked to both cognitive performance and metabolic demand (Goriounova & Mansvelder, 2019). These genes are implicated in various neuronal functions including synaptic function and plasticity, cell interactions and energy metabolism (Prabhakar et al., 2006; Whalen & Pollard, 2022) and relate to brain volume (Goriounova et al., 2018; Grasby et al., 2020).
The current phenotypic correlation between human intelligence and brain volume (BV) is estimated to be about .27 (Cox et al., 2019; Ritchie et al., 2015) (although earlier studies suggest a higher value) and the relationship may be due to shared genetic factors (Jansen et al., 2020). Especially in higher-order cortical areas, such as middle temporal gyrus (MTG), volume, cortical thickness and cortical activation were shown to be statistically associated with differences in intelligence quotient (IQ) scores in the modern human population (Choi et al, 2008). DMN supports internal attention and episodic memory retrieval, both critical for solving complex problems that require holding and manipulating abstract representations (Geary, 2024). Meta-analyses of voxel-based morphometry find that volumes of the frontal, parietal and temporal cortices each correlate with general intelligence at r ≈ 0.25–0.30 (≈ 6–9 % variance explained), very similar to the global brain-volume–IQ link of ~r = 0.24 (≈ 6 % variance). In particular, temporal-lobe volumes (including MTG) often reach r ≈ 0.28 in young-adult cohorts, but this local boost rarely exceeds r = 0.35 even in optimal samples. According to the Parieto-Frontal Integration Theory (P-FIT), fMRI activation in right BA 7 (parietal) and left DLPFC correlates with IQ in > 60–70 % of studies, with effect sizes around r = 0.30–0.40. Yet fMRI measures suffer from lower within-subject reliability (often ICC < 0.50), and are highly state-dependent, so they’re less consistent than structural biomarkers (Dubois et al., 2018; Elliott et al., 2020). Focusing on MTG or other association cortices can nudge the IQ-correlation up slightly, but doesn’t transform the predictive power – variance explained remains under 10 %. Moreover, region-specific structural and especially functional measures tend to have lower measurement reliability, making them no more dependable for forecasting individual differences in IQ, EA or SES than whole-brain volume.
Specific Genes of Interest
Common genetic variants contribute substantially to variation in measures of intelligence both within and between families, and rare variants, particularly rare protein-truncating variants (PTVs) in genes “intolerant of loss-of-function”, are associated with lower scores on intelligence tests as well as being implicated in rare developmental disorders. Here we discuss eight examples that are of particular interest because they suggest possible causal pathways based on previous research, and have been identified in more than one study. We recognize that each gene on its own likely has a minuscule effect and that additional research will require new analysis techniques that can elucidate complex systems of gene interactions.
1. CADM2 is one of the most significant common-variant loci in large GWAS of educational attainment (EA) and intelligence (Savage et al., 2018). It is also strongly implicated by GWAS and functional mutant mouse PheWAS studies in impulsive personality, with loss-of-function being less impulsive (Sanchez-Roige et al., 2023). It encodes Synaptic Cell Adhesion Molecule 2 (SynCAM2), a member of the immunoglobulin-superfamily that mediates homo- and heterophilic trans-synaptic adhesion. In neurons, it organizes excitatory synapses—promoting synapse formation, stabilizing presynaptic active zones, and tuning synaptic plasticity by regulating the number and strength of glutamatergic contacts (Frei & Stoeckli, 2017). However, like almost all individual common loci in polygenic traits, its per-allele effect on cognitive outcomes is very modest: median effect per risk allele corresponding to only about 1.7 weeks of additional schooling and at gene-level variance, aggregating all independent SNPs at the locus, ranging from about 0.07% up to about 3.0% of trait variance in large cohorts, with most estimates at the lower end of that range. Therefore, while CADM2 is genome-wide significant and biologically compelling—linking synaptic adhesion to cognitive variation—it is not a “major risk factor” in the clinical sense. Its influence may be one small piece of the highly polygenic architecture underlying IQ and educational attainment (J. J. Lee et al., 2018; Okbay et al., 2016, 2022)
2. Another similar locus is SLC39A8, the ZIP8 divalent cation transporter. Since it transports Zn2+, Mn2+ Fe2+ and Cd2+ metal ions, it illustrates the concept of pleiotropy, where a single gene can influence multiple, often seemingly disparate, biological pathways and illustrates how variants in this single gene can contribute to a range of complex conditions. Much focus is on its role as a manganese transporter which mediates Mn2+ uptake, essential in neurons, since Mn²⁺ is a cofactor for glycosyltransferases and for mitochondrial superoxide dismutase; SLC39A8 variants subtly alter Mn²⁺ homeostasis, impacting dendritic spine maintenance and synaptic plasticity. Neuronal SLC39A8 deficiency impairs cerebellar development and Slc39a8-inducible knockout mice showed that loss of SLC39A8 in brain micro vessels (but not in choroid plexus) reduces Mn2+ uptake into the brain by about 66%, identifying SLC39A8 in blood-brain barrier (BBB) endothelial cells as the key route for Mn2+ entry into the CNS. Manganese (Mn2+) is a critical cofactor for a surprisingly wide array of enzymes that underpin neurodevelopmental and synaptic functions, which explains why perturbing its uptake via SLC39A8 has outsized effects on cognition. Mn2+ is critical for many glycosyltransferases in the Golgi and therefore essential for proper protein folding (Choi et al., 2024; Lin et al., 2017; Pickrell et al., 2016).
3. Another of the polygenic predictors of IQ is FNBP1L, Formin-binding protein involved in orchestrating actin dynamics in developing neurons and actin cytoskeleton remodeling to promote dendritic spine formation and synaptic maturation, key to supporting efficient neurotransmission. Although FNBP1L stands out as the top single-gene hit in early GWAS (Benyamin et al., 2014; Davies et al., 2011), its per-allele impact is small: common variants at this locus account for <1% of trait variance in adults and only a few percent in children when combined in a polygenic score. This mirrors the highly polygenic architecture of intelligence, where hundreds of genes each contribute minute effects (Benyamin et al., 2014; Soderling et al., 2007).
4. These first three genes all encode proteins directly involved in the assembly of the synapse, reasonable as critical to overall neuronal function. Another family of IQ associated genes are the transcription factors, and none more clearly implicated than FOXP2 which acts as a master regulator of gene networks underlying neuronal maturation, synaptic plasticity and motor-learning circuits essential for language and higher-order cognition. It is highly expressed in layers 5/6 of cortex, basal ganglia (especially medium spiny neurons), cerebellar Purkinje cells and thalamic nuclei—regions critical for speech, motor sequencing and executive function (Vernes et al., 2011). Heterozygous FOXP2 mutations (e.g. R553H) cause developmental verbal dyspraxia accompanied by mild to moderate intellectual disability, directly demonstrating its impact on cognitive-motor integration (Lai et al., 2001). Further, common-variant analyses in multi-phenotype GWAS (educational attainment + cognitive performance) place FOXP2 among top loci whose polygenic scores explain about 7–10% of variance in cognitive measures (J. J. Lee et al., 2018; Savage et al., 2018). Like most transcription factors, FOXP2 directly binds and regulates hundreds of downstream targets, including synaptic genes, modulating neurite outgrowth, dendritic spine morphology and synaptic strength in excitatory circuits. Human-specific amino acid substitutions in FOXP2 alter its transcriptional activity in vitro and rewire gene-co-expression networks in developing cortex, suggesting an evolutionary role in shaping human cognitive capabilities (den Hoed et al., 2021; Konopka et al., 2009). Although it was initially surprising to find that the Neanderthal coding sequence was the same (Krause et al., 2007), it appears that there still are regulatory elements at the locus unique to modern man, leaving open the question of when language evolved (Maricic et al., 2013).
5. Almost in a class of its own is the fifth common variant gene, ATP1A1, which encodes the Na⁺/K⁺-ATPase, whose activity is essential for restoring resting membrane potential after each action potential, regulating cell volume, and driving secondary transport processes in all neurons. It is the single largest consumer of ATP in all mammals. Estimates for its share of resting metabolic energy is roughly 20–40% of basal (resting) metabolic rate, with human data indicating at least 20% of total energy expenditure is devoted to driving the pump, and in the brain approximately 50% of cerebral ATP turnover is used by Na⁺/K⁺-ATPase to maintain ionic gradients, figures underscore the enormous bioenergetic cost of maintaining ion homeostasis—especially in excitable tissues like the brain. Although expressed ubiquitously, the α1 isoform predominates at the plasma membrane of cortical and hippocampal neurons, where rapid firing demands robust Na⁺ extrusion and K⁺ uptake. Its role in IQ and fluid-intelligence is demonstrated by the recurrent Gly903Arg and other de novo variants that cause developmental delay, refractory epilepsy, and intellectual disability (Dohrn et al., 2024). In a recent fluid-intelligence GWAS (n > 450 K) by van den Berg and colleagues (van den Berg et al., 2025), ATP1A1 emerged among 26 genes whose aggregate rare variants (predicted loss-of-function or deleterious missense) are significantly associated with fluid-intelligence scores (FDR < 1%). Although no single common SNP in ATP1A1 attains the largest effect sizes seen in other pump subunits, aggregate common-variant signals at the ATP1A1 locus contribute to polygenic scores that explain a small but reproducible fraction of fluid-intelligence variance in UK Biobank.
6. CACNA1E is at genome-wide significance, unique among the ion channels. CACNA1E encodes Cav2.3 (R-type) voltage-gated calcium channels, which are expressed at high density in cortical pyramidal neuron dendrites and presynaptic terminals. These channels contribute disproportionately to dendritic calcium spikes and synaptic vesicle release, processes central to long-term potentiation, working memory, and excitatory drive. Because they directly regulate calcium entry into dendritic spines, they sit at a convergence point between electrical activity and transcriptional/metabolic signaling — precisely the domains repeatedly implicated in IQ. Its genetic effect size is unusually large for an ion channel. In contrast to sodium channel loci (SCN1A, SCN2A, SCN8A), which show clear enrichment but rarely reach genome-wide significance in common-variant GWAS, CACNA1E consistently emerges above threshold. This suggests that common variation at this locus exerts a more uniform and detectable impact on population-level traits like educational attainment, cognitive ability, or ASD risk.
Furthermore, CACNA1E shows a dual rare–common allele architecture. De novo LoF mutations in CACNA1E have been linked to severe developmental epileptic encephalopathy, while common regulatory alleles appear to shift dendritic excitability more subtly, nudging cognition or ASD liability. This “allelic series” makes it a powerful signal in large-scale GWAS. Also, its expression pattern aligns with human-specific cortical evolution. CACNA1E is strongly enriched in layer V pyramidal neurons and association cortices, including the prefrontal and parietal hubs of the default mode network (DMN) (van den Berg et al., 2025). These are the same networks expanded by human accelerated regions (HARs) and metabolically most vulnerable in ASD. That spatial specificity likely amplifies the impact of common variants, compared to broadly expressed ion channels whose effects diffuse across multiple neuronal types. This evolutionary constraint strengthens the signal. CACNA1E is highly constrained against LoF variation in the gnomAD database, the largest publicly available resource of aggregated human genetic variation, meaning even mild common variants are more likely to be under selection and thus detectable in association studies.
Similarly, CACNA1A encodes the α₁A subunit of the P/Q-type voltage-gated Ca²⁺ channel (Cav2.1), and also contributes to human cognitive ability primarily through rare coding variants whose burden is significantly associated with verbal-numerical reasoning (a proxy for IQ) in exome-wide analyses of the UK Biobank study ~455,000 individuals (C. Y. Chen et al., 2023). Its protein-truncating and damaging missense variants are one of only eight genes to reach exome-wide significance (P < 3.2 × 10⁻⁶) for verbal-numerical reasoning scores, with variant carriers showing lower performance. This channel drives presynaptic Ca²⁺ influx at excitatory synapses and its Ca²⁺ entry is essential for vesicular neurotransmitter release, dendritic Ca²⁺ signaling, and the induction of synaptic plasticity—all cellular substrates of learning and memory (Adams et al., 2010). Loss or missense disruption of these channels slows synaptic transmission and impairs long-term potentiation, leading to measurable deficits in problem-solving and reasoning (Damaj et al., 2015).
7. A number of other voltage-gated Ca²⁺ channel subunits are also implicated in IQ. In the UK Biobank exome dataset (n≈450 000) (Van Hout et al., 2020), aggregate rare (protein-truncating+ deleterious missense) variants in CACNA1C have been associated exome-wide with lower verbal-numerical reasoning scores (P < 3 × 10⁻⁶), ranking it among the top handful of ion-channel genes whose rare-variant burden impacts fluid-intelligence measures. This finding highlights CACNA1C as not only a psychiatric risk gene but also a core determinant of normal-range cognitive function, consistent with its role in shaping dendritic calcium transients and gene transcription programs critical for synaptic plasticity. At the common-variant level, the SNP rs1006737, located in intron 3, is one of the most widely replicated psychiatric risk alleles. The “A” risk allele is associated with subtle but consistent cognitive effects, including reduced delayed-memory performance and weaker executive-function scores in both healthy adults and patients with mood or psychotic disorders. These cognitive signatures suggest that CACNA1C variation perturbs calcium-dependent regulation of prefrontal and hippocampal circuits, producing a spectrum of phenotypic outcomes from modest cognitive differences in the general population to profound deficits in psychiatric disease. Meta-analyses and candidate-gene studies show that A-carriers perform worse on verbal-memory tasks (effect sizes ~0.2 SD), and neuroimaging reveals lower hippocampal integrity and altered medial prefrontal-cortex morphology in carriers (M. Chen et al., 2022). While common alleles in CACNA1C confer modest, polygenic effects on memory and executive tasks, rare coding mutations yield dramatic cognitive impairment (Splawski et al., 2004). Together, these data—from monogenic syndromes, candidate-SNP associations, and large-scale exome analyses—establish CACNA1C as a gene whose perturbation across the allele-frequency spectrum influences human IQ and related cognitive traits.
8. The single most common locus identified in GWAS of ASD is SCN2A (Koko et al., 2025; Satterstrom et al., 2020) and it is also strongly implicated in GWAS of IQ/EA/SES (DDDS, 2017) (Deciphering Developmental Disorders Study). It encodes the voltage-activated sodium channel Nav1.2, which mediates action-potential initiation and dendritic excitability, and rare de novo truncating and missense loss of function (LoF) alleles contribute to both phenotypes. The DDD Study sequenced 4,293 trios and found 19 individuals with de novo SCN2A loss-of-function variants, placing SCN2A among the top 94 genes with genome-wide significant enrichment of damaging de novo mutations in children with severe neurodevelopmental delay and intellectual disability (estimated incidence 1/47,000–1/100,000 births). Heterozygous Scn2a⁺/⁻ mice (mimicking human LoF) show impaired spatial learning, reduced long-term potentiation, and decreased perirhinal-cortex–driven hippocampal plasticity—demonstrating that even a 50% reduction in Nav1.2 seriously compromises learning and memory circuits.
Intriguingly, it is predominantly expressed in excitatory pyramidal neurons of the cortex and hippocampus, where it localizes to axons and dendrites to support back-propagating depolarizations. This positioning allows Nav1.2 to shape the initiation and fidelity of action potentials in the axon initial segment and to sustain their back-propagation into dendrites, a process essential for coupling synaptic input with somatic output (Gidon et al., 2020; Spratt et al., 2019). By amplifying depolarizing signals within dendritic trees, SCN2A enables the calcium influx required for activity-dependent gene expression and long-term synaptic plasticity. Thus, it serves as a critical biophysical link between fast spike conduction and slower adaptive processes such as learning and memory. Loss-of-function variants reduce excitability and impair dendritic integration, leading to deficits in associative plasticity and cognitive performance, while gain-of-function mutations can destabilize firing patterns, producing epileptiform activity (Bender & Trussell, 2012).
This dual role—as both a gatekeeper of normal excitatory transmission and a potential driver of pathological network hyperexcitability—explains why SCN2A mutations span a clinical spectrum from intellectual disability and autism to severe infantile epilepsies (Ben-Shalom et al., 2017). Its close relative SCN1A, on the other hand, is primarily enriched in fast-spiking GABAergic interneurons in cortex and hippocampus, with Nav1.1 concentrated at their axon initial segments (AIS) or hillock (Hedrich et al., 2014). By being clustered at the AIS, Nav1.1 channels ensure the rapid, high-frequency firing of inhibitory interneurons—particularly parvalbumin+ basket cells—allowing them to exert tight temporal control over pyramidal neuron excitability. This strategic localization enables interneurons to synchronize cortical networks through gamma-band oscillations, a rhythm essential for working memory, sensory binding, and higher cognitive functions. When Nav1.1 function is reduced, as in SCN1A haploinsufficiency, interneurons lose their ability to fire reliably at high frequency, tipping the excitatory–inhibitory balance toward hyperexcitability. The resulting breakdown in network synchronization explains both the seizure susceptibility and the cognitive impairments that characterize Dravet syndrome— a rare, severe form of epilepsy that typically begins in the first year of life (Yu et al., 2006).
On the other hand, it is one of the most common loci identified as “only missense/never LoF” in ASD (Satterstrom et al., 2020), where mutations are localized to the calmodulin-binding domain, suggesting a weaker down-regulation of the channel. This clustering points to a distinct pathogenic mechanism: rather than abolishing Nav1.1 function, ASD-associated alleles appear to subtly alter channel gating and calcium-dependent modulation (Schmunk & Gargus, 2013). Such variants likely reduce interneuron excitability just enough to shift the excitatory–inhibitory balance, without crossing the catastrophic threshold that produces epileptic encephalopathy. This intermediate phenotype may account for why such SCN1A variants are enriched in ASD cohorts, where cognitive and social deficits predominate but seizures are less universal or severe. The location of these alleles in regulatory regions of the channel further suggests they interfere with activity-dependent feedback, potentially impairing the fine-tuning of inhibitory tone during developmentally critical windows of circuit formation. In this way, SCN1A provides a rare example of an ion channel gene where allelic series—from haploinsufficiency to weak functional perturbation—map onto a clinical continuum from Dravet syndrome to autism. Therefore, rare loss-of-function mutations in SCN1A produce a clinical syndrome marked by profound cognitive impairment, and interneuron-specific Scn1a⁺/⁻ mice recapitulate memory and learning deficits, while weak disruption-of-function alleles are associated with ASD—together providing the clearest functional link between SCN1A loss of function and loss of GABAergic interneuron function leading to excitation/inhibition imbalance through loss of inhibitory tone and impact on human IQ (Léna & Mantegazza, 2019).
Summary of specific genes: To date, intelligence (g) has proven extremely polygenic—hundreds of loci reach genome-wide significance in single studies, but only a handful replicate across independent GWAS, and those two, SLC39A8 and CADM2, were discussed above. Most intelligence-associated SNPs explain vanishingly small fractions of variance, making replication across cohorts difficult. By contrast, alleles in SLC39A8 and CADM2 reproducibly shift cognitive scores across independent studies, indicating that their effect sizes are both larger and more stable than the polygenic background. Of the hundreds of loci identified in each study— (J. J. Lee et al., 2018) reporting 1,271 EA loci, (Savage et al., 2018) 205 “g” loci (mapping to 1,016 genes), and (Davies et al., 2018) 148 CP loci—the only two genes consistently significant and replicated across all three are CADM2 and SLC39A8. Both converge on core synaptic biology: CADM2 via trans-synaptic adhesion complexes that scaffold excitatory contacts, and SLC39A8 by regulating manganese homeostasis critical for enzyme cofactors in glycosylation and oxidative stress defense. Because these loci influence fundamental processes (metal transport, synaptic connectivity), even modest allelic differences are under selection, leading to common variants of relatively large effect persisting in populations — a recipe for reproducibility across GWAS. They both sit at the intersection of common-variant and rare-disease biology. Both genes are highly constrained: rare loss-of-function mutations are linked to neurodevelopmental disorders, while common variants subtly modulate cognitive traits in the general population. This rare–common continuum makes their signals unusually replicable.
What Might Polygenic Scores Tell Us?
While polygenic scores for intelligence (IQ), cognitive performance (CP), and educational attainment (EA) are increasingly used, it is not known which aspects of general intelligence are reflected and to what extent. Genç et al (2021) show that IQ-PGS, CP-PGS, and EA-PGS do not predict every form of cognitive ability equally well and found for all three PGS for the whole group and separated for the sexes the pattern that all PGS had a high predictive power for interindividual differences in general, verbal, and numerical intelligence. In contrast, memory was only weakly associated. More specifically, these measures are more strongly associated with crystallized cognitive abilities compared to fluid abilities, contrary to some classical models which assumed that crystallized abilities would be less influenced by genetics but more impacted by environmental factors, like education. Nevertheless, recent evidence from a meta-analytic twin study showed higher heritability estimates for crystallized compared to fluid abilities (Kan et al., 2013), speculating that these findings could be explained in terms of genotype-environment covariance, since crystallized abilities depends on fluid abilities, and that high achievers are more likely to end up in cognitively demanding environments which facilitate the initial genetic predisposition and thus the further development of a wide range of knowledge and skills (Genc et al., 2021). It may also be the case that the higher association is due in part to crystallized measures generally being more reliable.
Remarkably of the 1,257 genome-wide significant personality loci (Schwaba et al., 2025) and the 205 “g” loci (Savage et al., 2018) or the 1,271 EA loci (J. J. Lee et al., 2018), the same two genes, CADM2 and SLC39A8 are the only ones to reach genome-wide significance (p < 5×10⁻⁸) in all. This pattern—moderate trait-level overlap (especially for openness and conscientiousness; the former positively correlated with intelligence) but minimal direct locus-sharing—illustrates that personality and intelligence share broad polygenic influences on neurodevelopment and synaptic function, yet most of their individual risk variants remain distinct.
No other single gene consistently meets genome-wide significance and independent replication, a high standard for the validity of the discovery. However, meta-analyses nominate dozens more loci—such as the three transcription factors MEF2C, FOXO6 and TCF4, and especially the axon guidance receptors DCC and ROBO. Their lead SNPs typically fail to replicate at p < 5 × 10⁻⁸ in a second, well-powered cohort studies, but each exerts only a whisper of influence on cognitive traits. Their true value lies in pointing toward neurodevelopmental pathways (axon guidance, synapse formation, transcriptional control), rather than serving as “major risk factors” in isolation. This is a critical point about the value of GWAS results.
These three transcription factors each have multi-level support for roles in human intelligence: MEF2C by monogenic haploinsufficiency and common-variant regulatory effects (Ali et al., 2024; Le Meur et al., 2010); FOXO6 by gain-of-function in mouse models revealing synaptic- and memory-related gene programs (Salih et al., 2012); and TCF4 by both rare, large-effect mutations causing severe intellectual disability and common alleles affecting reasoning in clinical cohorts (Albanna et al., 2014; Amiel et al., 2007). MEF2C controls expression of proteins involved in mitochondrial energy pathways and ATP production as well as synapse remodeling (Ali et al., 2024); FOXO6 modulates the expression of proteins involved in the insulin/IGF-PI3K and p53 signaling pathways which integrate metabolic cues with synaptic plasticity and neuronal survival, a gene network enriched for glutamate-receptor signaling, synapse-formation factors, and dendritic-spine regulators, deficits which underlie its memory-consolidation phenotype (Salih et al., 2012). TCF4 regulates the expression of proteins in the Wnt/β-catenin/ SOX axis with loss-of-function mutations impairing Wnt/β-catenin signaling and down-regulating SOX transcription factors, as well as SMADs, IGF2/IGFBPs and caspases/NF-κB that are all essential for neural-progenitor proliferation (Forrest et al., 2013; Papes et al., 2022).
DCC and ROBO1 are both implicated in large GWAS for EA (J. J. Lee et al., 2018; Okbay et al., 2016). While the evidence suggests these genes being significant, GWAS findings explain only a small portion (~12-16%) of variance in educational attainment. DCC acts as a receptor for netrin-1, crucial for axon guidance during neural development, ensuring proper wiring of neural circuits, which could influence learning and cognitive processes. It is also associated with brain volume, a correlate of IQ (Morgunova et al., 2020). ROBO1 is a receptor for slit proteins, essential for axon repulsion and neural circuit formation, associated with dyslexia and phonological processing in family-based analyses and playing a key role in establishing brain networks that support intelligence and cognitive functions (J. J. Lee et al., 2018; Okbay et al., 2016; Tran et al., 2014).
Diet and other metabolic considerations relevant to intelligence
In addition to the genes and their expression profile already discussed, other biological influences are of molecular interest including diet, PUFAs, DHA, MFSD2A, and mitochondria (Gargus, 2025).
Diet-related considerations: The adult human brain accounts for only 2% of body mass, but requires >20% of basal metabolic energy, a value that approaches 60% in the neonate (Kuzawa et al., 2014). These energetic demands could not be supported amid near-starvation conditions of the Ice Age, restricting early hominid brain volume. In the Marine Isotope Stage 6 (MIS6), dramatic Ice Age climate shifts made much of the land uninhabitable, depositing huge glaciers onto the lands. However, this produced falling sea levels that exposed rich mussel beds on the narrow inhabitable zone at the South African tip (Marean, 2011, 2015, 2016). Suddenly, this dependable nutrient-rich marine food source amid otherwise near-starvation Ice Age conditions—and especially a source rich in terrestrially-rare long-chain polyunsaturated fatty acids (PUFAs), like docosahexaenoic acid (DHA), required for synapse formation (Brenna & Carlson, 2014)—enabled fetuses carrying mutations that caused sustained brain expansion to no longer be lethal because of an unsustainable energetic load.
Polyunsaturated fatty acids (PUFAs): PUFAs have been evolutionarily-selected as signal transducing lipids highly enriched in neuronal synaptic membranes, particularly in lipid rafts surrounding calcium channels (Crawford et al., 2013). Independent evidence suggests that endoplasmic reticulum (ER) membranes, particularly at “mitochondria-associated membrane” (MAM) domains, are modulated by PUFA content, affecting calcium channel gating and inter-organelle signaling (Arruda & Hotamisligil, 2015; Fujimoto & Hayashi, 2011; Innis, 2008; Paillusson et al., 2016). MAMs are critical regions of macromolecular assembly at contacts between the two organelles in all cells, specifically where the ER’s inositol 1,4,5-trisphosphate receptors (ITPRs) and mitochondrial voltage-dependent anion-selective channel (VDAC) pores, form a direct calcium pathway that conducts triggered release of ER intracellular calcium stores directly into the mitochondrial matrix. There calcium ions set the rate of the mitochondrial tricarboxylic acid cycle (TCA) via its three calcium-regulated dehydrogenases. It is the TCA-generated reduced NADH and FADH2 that chemiosmotically power proton flux through the respiratory chain of the inner mitochondrial membrane to drive ATP production. With the ER serving as the sensor of cellular ATP consumption and stress, physiologically, this circuit is a homeostatic mechanism that allows ATP production to match ATP consumption, orchestrating cell survival and synaptic plasticity (Cárdenas et al., 2010; R. L. Nguyen et al., 2018). Perturbations of this essential circuit are critically vulnerable to pathogenesis.
Docosahexaenoic acid (DHA): PUFAs are required in abundance during neurogenesis (Bazinet & Layé, 2014; Innis, 2007; Madore et al., 2020). During the final trimester of human pregnancy fetal DHA accretion reaches its peak in the last five weeks of gestation. In fact, the amount of DHA deposited in the brain between ~36–40 weeks increases exponentially and is about equal to the total accumulated in the first 35 weeks (Hachem et al., 2016; Martínez & Mougan, 1998). This late-gestation spike in DHA uptake coincides with a period of rapid brain growth and maturation as the blood-brain barrier (BBB) and neural tissues develop intensive DHA demand. By around 35 weeks, the fetal BBB is functionally mature enough to support high rates of nutrient transport, and DHA uptake into the brain accelerates markedly. This developmental timing helps explain why infants born prematurely (before ~37 weeks) miss out on the peak in utero DHA transfer—a gap associated with suboptimal neurodevelopmental outcomes (S. L. Smith & Rouse, 2017).
After birth, the infant’s brain continues to avidly accumulate DHA, though the source shifts to dietary intake (breast milk or formula). Brain DHA uptake remains high throughout the first two years of life, a phase of rapid postnatal brain development. Quantitative analyses have shown a massive increase (on the order of 20–30×) in total brain DHA content from birth until about 24 months of age (Hachem et al., 2016). This indicates that the infant BBB still transports DHA at an accelerated rate during infancy to meet the needs of ongoing neurodevelopment. Autopsy studies and lipid profiles confirm that DHA accrues rapidly in the human brain from late gestation through the first 18–24 months, after which the rate of accretion tapers off significantly. By approximately 2 years of age, a child’s brain has achieved >80% of its adult size and fatty acid composition, and the previously high DHA uptake starts to decline to a steadier, maintenance level (S. L. Smith & Rouse, 2017).
PUFAs and membrane dynamics affecting IP₃ signaling
PUFAs are integral components of cellular membranes, where they influence membrane fluidity and the organization of lipid rafts—microdomains that compartmentalize cellular processes (Paillusson et al., 2016; Vance, 2014). By altering the lipid environment, PUFAs can modulate the localization and function of membrane-associated proteins, including IP₃Rs. Changes in membrane composition due to PUFA incorporation can affect the efficiency of IP₃-mediated Ca²⁺ release by influencing receptor conformation and interactions with signaling partners (Katona et al., 2022).
PUFAs as precursors to bioactive lipids influencing IP₃ signaling
PUFAs serve as precursors to various bioactive lipid mediators, such as diacylglycerol (DAG) and arachidonic acid (AA), which are generated through phospholipase C (PLC) activity following receptor stimulation. These lipid mediators can modulate IP₃ signaling pathways by influencing the activity of protein kinase C (PKC) and other downstream effectors, thereby affecting IP₃R sensitivity and Ca²⁺ release. Additionally, metabolites derived from PUFAs can directly or indirectly interact with IP₃Rs, further modulating their function (Katona et al., 2022).
In summary, PUFAs contribute to IP₃ signaling and IP₃R function through multiple mechanisms: by serving as substrates for palmitoylation essential for IP₃R stability, by modulating membrane properties that influence receptor behavior, and by generating lipid mediators that affect downstream signaling pathways.
Major Facilitator Superfamily Domain-containing protein 2A (MFSD2A): MFSD2A is the key, highly conserved transporter that enables DHA to cross the blood-brain barrier. MFSD2A is highly expressed in the endothelial cells of the BBB’s microvasculature, and it mediates the uptake of DHA from blood into the brain (L. N. Nguyen et al., 2014). This mechanism is crucial for neurodevelopment: MFSD2A’s role is illustrated by rare loss-of-function mutations in the human MFSD2A gene, which cause severe microcephaly and neurodevelopmental impairment, directly demonstrating the impact of PUFA supply on the potential for brain expansion (Wood et al., 2021). These cases provide direct evidence that in humans, normal brain growth in late fetal and early postnatal life depends on massive increases in PUFA across the blood brain barrier (Guemez-Gamboa et al., 2015). This very late timepoint in development suggests ontogeny recapitulating phylogeny (Gould, 1977), and that this very active PUFA transport may reflect its selection during the most recent evolutionary pressures associated with metabolically-intensive, expanded, synapse-rich cortical regions like the precuneus.
From an energetic standpoint, the modern human brain continues to consume the vast portion of resting metabolic output, a disproportion most extreme in early development, when the precuneus is still undergoing myelination and synaptogenesis. This energetic bottleneck likely constrained brain growth throughout hominid evolution (Harris et al., 2012). The sudden ecological access to high-quality nutrition and dietary lipids (e.g., DHA) during the Marine Isotope Stage 6 (MIS6) foraging revolution (Marean, 2010) may have relaxed these constraints, enabling a rapid expansion of energy-hungry networks like the DMN—supported by the emergence of HAR-driven regulatory innovations.
Mitochondria: Mitochondria play a critical role in fueling the brain’s highly oxidative-dependent energy needs and may be related to cognition and IQ scores (Geary, 2021; Shatalina et al., 2024; Xia et al., 2025). The mitochondria’s oxidative metabolism of glucose, its normal energy source, provides 16 times the ATP of anaerobic metabolism. A few minutes of anoxia causes a rapid shutdown of the brain, leading to coma and death (Howarth et al., 2012). Even brief fasting illustrates the brain’s high sensitivity to its extraordinary energy demands. At the onset of fasting, blood glucose is quickly consumed, and the liver begins to release glucose from glycogen stores to temporarily support the brain while other tissues switch to oxidizing long-chain fatty acids. However, these fats cannot cross the BBB, so again a special process is developed to support the brain when glycogen’s glucose is gone. Ketone bodies, acetoacetate and β-hydroxybutyrate, which can travel on monocarboxylate transporters across the BBB, are produced for the brain by the liver to be oxidized by the brain’s mitochondria (N. J. Jensen et al., 2020). While other primates utilize ketone bodies to a limited extent, they still largely rely on glucose. The evolution of the modern human brain not only greatly enhanced the capability of ketone body production, but also generated large fat supplies in the fetus, unique among the primates (Cunnane & Crawford, 2014), to assure a secure supply of lipids for newborn brain growth and its energy supply.
In Autism Spectrum Disorder (ASD), for example, mild mitochondrial dysfunction is a common finding, evidenced by impaired oxidative phosphorylation, elevated lactate and alanine levels, carnitine deficiency, abnormal reactive oxygen species (ROS) and altered calcium homeostasis, but much less frequently mitochondrial DNA mutations (Filipek et al., 2004; Gargus & Imtiaz, 2008; Hagihara et al., 2024; Palmieri et al., 2010; Rossignol & Frye, 2012; M. Smith et al., 2012). These are largely post-translational epigenetic functional mitochondrial defects, and they are particularly relevant in the context of energy-hungry brain regions like the cortical “social brain” and precuneus, areas amongst the brain structures displaying the highest resting metabolic rates and genetically- vulnerable to ASD (Gargus, 2025).
A note on possible interactions with culture and Social Economic Status (SES)
There is a long running issue about how genetic influences may interact with SES and other cultural variables. Most recently, Abdellaoui and colleagues argue, socioeconomic status (SES)—far from being a purely social construct—acts as an emergent selective regime (Abdellaoui et al., 2025). Through gene–culture coevolution, SES influences mating patterns, cognitive development opportunities, and reproductive outcomes. This can produce second-order genetic effects that shape allele frequencies of traits linked to intelligence, reinforcing the polygenic foundation uncovered by Oxley and colleagues (Oxley et al., 2024) and by (J. J. Lee et al., 2018); see figure 1.Their perspective, however, is largely conceptual and stops short of quantifying actual selection coefficients on intelligence-linked alleles. The broader theory of gene–culture coevolution posits feedback loops between cultural practices and genetic change e.g. (Cavalli-Sforza & Feldman, 1981), and Abdellaoui et al. invoke how SES-driven cultural environments could reshape gene frequencies over generations. That said, the paper does not present formal gene–culture models or direct data on cultural selection dynamics; it remains a compelling hypothesis rather than a fully solved empirical demonstration.
As for “second-order genetic effects” on allele frequencies, while it’s plausible that SES-associated selection pressures tweak allele frequencies for intelligence-related loci, direct evidence is scarce. Contemporary GWAS (and family-based studies) mainly capture standing variation in polygenic scores but have not yet detected clear signals of recent selection acting specifically on those scores. Lee et al. (2018) identified over 1,200 genome-wide significant loci for educational attainment, indicating its highly polygenic architecture. Oxley et al. (2024) meta-analyzed polygenic-score prediction across multiple cohorts and found that polygenic scores explain roughly 27 % of variance in IQ, further indicating a broad polygenic basis for intelligence.
So, the notion that SES itself can function as a selective regime is theoretically justified and conceptually supported by Abdellaoui et al., but empirical quantification of how much allele frequencies for intelligence traits have shifted due to SES is still lacking. Likewise, invoking gene–culture coevolution enriches the narrative but remains to be formalized and tested against data. Finally, Lee et al. (2018) and Oxley et al. (2024) establish the polygenic nature of educational attainment and IQ, respectively—but linking those GWAS hits back to SES-driven selection will require more direct evolutionary-genomic evidence; for example, see (Wang et al., 2025).
Conclusion: Putting it All Together
The findings we have reviewed support the proposition that individual differences in intelligence in the population reflect the tradeoff between the historically recent rapid evolving demands of our brains and the metabolic systems that support them. Our social, symbol-using enlarged energy-hungry brains gave our direct ancestors language, art, weapons, and culture, all allowing our small cohort of relatives to “emerge from a narrow bottleneck in Africa”, and cover the entire globe (Marean, 2010, 2016) with our current tiny pool of genetic diversity (Prado-Martinez et al., 2013), but concomitantly creating a vulnerability from its huge energetic burden. Diversity in the robustness of support to the delicate balance of energy supply during brain development, whether due to mitochondrial issues, calcium signaling problems, or environmental stressors, may cause these newly-evolved energy-demanding networks to tip the IQ scale. This theory does not suggest that evolution “caused” intellectual disability. Instead, it offers a framework for understanding why certain children may be more vulnerable, especially when their developing brains do not receive the necessary energy support they need. The other side of the vulnerability coin is whether individuals might benefit from a “better” balance of energy demanding brain networks that might maximize or enhance intelligence. The challenge is to empirically define “better.”
Few intelligence researchers have the necessary molecular genetics training to undertake testing hypotheses about the mechanisms of brain energy and their possible relationships to cognitive abilities, especially the g-factor. Many researchers around the world, however, are already conducting molecular neuroscience studies of learning and memory on patients with dementia, schizophrenia, depression, and autism spectrum disorder. We hope the same research designs, techniques, and analyses can be applied to non-patient samples of high, average, and low scorers on psychometric tests of intelligence or on tests of specific cognitive abilities; PGS can be an important independent variable or a covariant. Such research could be the start of a new phase of intelligence research that in the long run may lead to insights for the ultimate goal of enhancement for individuals.
For example, here are some questions to consider:
-
Can PGS predict which infants might benefit cognitively from increased PUFA or other lipids?
-
Are any types of intellectual disability known to be lipid related?
-
Do embryos with high PGS for intelligence have any interesting lipidomic or metabolic profiles?
-
Are there any mice knock outs with lipid disruption and cognitive problems? See Gargus, 2025 for such a potential spontaneous mutant.
-
What are any molecular pathways associated with cognitive decline or with brain injuries that effect cognition?
-
Are there any measures of mitochondria that covary with PGS for EA or IQ? See (Mosharov et al., 2025; Xia et al., 2025).
-
Does damage limited to the precuneus (like stroke) cause specific or general cognitive issues that can be predicted by PGS?
-
What brain networks are revealed if a connectome seed is placed in the precuneus and do they vary according to PGS?
-
In addition to the precuneus, what other brain area connections might reveal relationships that imply causal mechanisms for intelligence? See (Ralph et al., 2017; Ünal et al., 2025).
-
Do any of the genes associated with intelligence result in trade-offs or costs in other systems?
None of these questions were generated by AI but we had some fun asking various AI programs to suggest research hypotheses and experiments to test ways to use molecular biology findings from learning and memory research to enhance intelligence. For now, these were merely exercises and we encourage researchers to carefully test their own prompts for potential AI help. We note that AI advances have explosive requirements for enormous supplies of electrical energy and this may evolve to migration to special ecological niches of low-cost energy. It is tempting to draw a conceptual parallel to the energy demands of the evolution of human general intelligence. Hopefully any parallel ends without the prior inexorable competitive-megafuna extinctions.
Will the quest for General Artificial Intelligence ever overlap with the search for molecular ways to enhance human intelligence or will GAI success (and control) depend ultimately on enhancement of human g? Fifty years ago, there was heated controversy about whether genes had anything at all to do with intelligence differences among people or even with diseases like schizophrenia or autism. Perhaps it will take another fifty years, but based on history, scientific advances seem inevitable even on the most complex and contentious of questions.